publications
2024
- bioRxivPREPRINT: Early founder effects have determined parental population structure in the Faroe IslandsA.E. Mann, E. Magnussen , and C.R. TillquistbioRxiv, 2024
The Faroe Islands are a small archipelago located in the North Atlantic likely colonized by a small group of founders sometime between 50 and 300 CE. Post colonization, the Faroese people have been largely isolated from admixture with mainland and other island populations in the region. As such, the initial founder effect and subsequent genetic drift are likely major contributors to the modern genetic diversity found among the Faroese. In this study, we assess the utility of Y-chromosomal microsatellites to detect founder effect in the Faroe Islands through the construction of haplotype networks and a novel empirical method, mutational distance from modal haplotype histograms (MDM), for the visualization and evaluation of population bottlenecks. We compared samples from the Faroe Islands and Iceland to possible regional source populations and documented a loss of diversity associated with founder events. Additionally, within-haplogroup diversity statistics reveals lower haplotype diversity and richness within both the Faroe Islands and Iceland, consistent with a small founder population colonizing both regions. However, in the within-haplogroup networks, the Faroe Islands are found within the larger set of potential source populations while Iceland is consistently found on isolated branches. Moreover, comparisons of within-haplogroup MDM histograms document a clear founder signal in the Faroes and Iceland, but the strength of this signal is haplogroup-dependent which may be indicative of more recent admixture or other demographic processes. The results of the current study and lack of conformity between Icelandic and Faroese haplotypes implies that the two populations were founded by different paternal gene pools and there is no detectable post-founder admixture between the two groups.
- Research SquarePREPRINT: HIV Infection and Exposure Increases Cariogenic Taxa, Reduces Taxonomic Turnover, and Homogenizes Spatial Differentiation for the Supragingival MicrobiomeA.E. Mann, C. Aumend , S. Crull , and 13 more authorsResearch Square, 2024
The oral microbiome comprises distinct microbial communities that colonize diverse ecological niches across the oral cavity, the composition of which are influenced by nutrient and substrate availability, host genetics, diet, behavior, age, and other diverse host and environmental factors. Unlike other densely populated human-associated microbial ecosystems (e.g., gut, urogenital), the oral microbiome is regularly and directly exposed to the external environment and is therefore likely less stable over time. Cross sectional studies of the oral microbiome capture a glimpse of this temporal dynamism, yet a full appreciation of the relative stability, robusticity, and spatial structure of the oral environment is necessary to understand the role of microbial communities in promoting health or disease. Here we investigate the spatial and temporal stability of the oral microbiome over three sampling time points in the context of HIV infection and exposure. Individual teeth were sampled from a cohort of 565 Nigerian children with varying levels of tooth decay severity (i.e., caries disease). We collected 1,960 supragingival plaque samples and characterized the oral microbiome using a metataxonomic approach targeting an approximately 478 bp region of the bacterial rpoC gene. We found that both infection and exposure to HIV have significant effects on the stability of the supragingival plaque microbiome at both the spatial and temporal scale. Specifically, we detect (1) significantly lower taxonomic turnover of the oral community among exposed and infected children compared to unexposed children, (2) we find that HIV infection homogenizes the oral community across the anterior and posterior dentition, and (3) that impaired immunity (i.e., low CD4 count) and low taxonomic turnover over time in children living with HIV is associated with higher frequency of cariogenic taxa including Streptococcus mutans. Our results document substantial community fluctuations over time in children unexposed to HIV independent of oral health status. This suggests that the oral community, under typical conditions, rapidly adapts to environmental perturbations to maintain homeostasis and that long-term taxonomic rigidity is a signal of community dysfunction, potentially leading to a higher incidence of oral disease including caries.
- Sci. Rep.Metagenomic and paleopathological analyses of a historic documented collection explore ancient dental calculus as a diagnostic toolR.M. Austin , T.P. Honap , A.E. Mann, and 5 more authorsScientific Reports, 2024
Dental calculus is a microbial biofilm that contains biomolecules from oral commensals and pathogens, including those potentially related to cause of death (CoD). To assess the utility of calculus as a diagnostically informative substrate, in conjunction with paleopathological analysis, calculus samples from 39 individuals in the Smithsonian Institution’s Robert J. Terry Collection with CoDs of either syphilis or tuberculosis were assessed via shotgun metagenomic sequencing for the presence of Treponema pallidum subsp. pallidum and Mycobacterium tuberculosis complex (MTBC) DNA. Paleopathological analysis revealed that frequencies of skeletal lesions associated with these diseases were partially inconsistent with diagnostic criteria. Although recovery of T. p. pallidum DNA from individuals with a syphilis CoD was elusive, MTBC DNA was identified in at least one individual with a tuberculosis CoD. The authenticity of MTBC DNA was confirmed using targeted quantitative PCR assays, MTBC genome enrichment, and in silico bioinformatic analyses; however, the lineage of the MTBC strain present could not be determined. Overall, our study highlights the utility of dental calculus for molecular detection of tuberculosis in the archaeological record and underscores the effect of museum preparation techniques and extensive handling on pathogen DNA preservation in skeletal collections.
- Micro SpectrumHeterogeneous lineage-specific arginine deiminase expression within dental microbiome speciesA.E. Mann, B. Chakraborty , L.M. O’Connell , and 3 more authorsMicrobiology Spectrum, 2024
Arginine catabolism by the bacterial arginine deiminase system (ADS) has anticariogenic properties through the production of ammonia, which modulates the pH of the oral environment. Given the potential protective capacity of the ADS pathway, the exploitation of ADS-competent oral microbes through pre- or probiotic applications is a promising therapeutic target to prevent tooth decay. To date, most investigations of the ADS in the oral cavity and its relation to caries have focused on indirect measures of activity or on specific bacterial groups, yet the pervasiveness and rate of expression of the ADS operon in diverse mixed microbial communities in oral health and disease remain an open question. Here, we use a multivariate approach, combining ultra-deep metatranscriptomic sequencing with paired metataxonomic and in vitro citrulline quantification to characterize the microbial community and ADS operon expression in healthy and late-stage cavitated teeth. While ADS activity is higher in healthy teeth, we identify multiple bacterial lineages with upregulated ADS activity on cavitated teeth that are distinct from those found on healthy teeth using both reference-based mapping and de novo assembly methods. Our dual metataxonomic and metatranscriptomic approach demonstrates the importance of species abundance for gene expression data interpretation and that patterns of differential expression can be skewed by low-abundance groups. Finally, we identify several potential candidate probiotic bacterial lineages within species that may be useful therapeutic targets for the prevention of tooth decay and propose that the development of a strain-specific, mixed-microbial probiotic may be a beneficial approach given the heterogeneity of taxa identified here across health groups.
2023
- MicroorgA synthetic formula amino acid diet leads to microbiome dysbiosis, reduced colon length, inflammation, and altered locomoter activity in C57BL/6J miceV.J. Mancilla , P.N. Braden-Kuhle , K.N. Brice , and 8 more authorsMicroorganisms, 2023
Although a typical treatment option for metabolic disorders is synthetically derived formula diets, the effects have not been extensively studied, and may render other health consequences. Dietary fiber is essential to protect the gut from opportunistic pathogens and increase the population of commensal bacteria in the colon, which is the most densely populated area for microbiota in the human body. Previous research has demonstrated that diets high in fiber are crucial to human health, as they increase microbial diversity and short chain fatty acid production and allow for proper maintenance of gut tissue. Notably, evidence suggests that high-fiber diets improve cognition and provide neuroprotection, as the gut and brain are connected by the gut-brain axis. Conversely, previous studies in both human subjects and animals have shown that low-fiber diets are associated with decreased gut bacterial diversity, decreased short chain fatty acid production, increased gastrointestinal distress, and increased cognitive dysfunction and anxiety-like behavior. In the current, multidisciplinary study, we examined the effects of a synthetically derived, low-fiber, amino acid diet on anxiety-like behavior, locomotor activity, cognition, the gut microbiome, cytokines, colon length, and short-chain fatty acid production in male C57BL/6J mice. Mice were assigned to one of two diet conditions at post-natal day 21: a standard rodent diet or a synthetic diet analogous to the standard rodent diet, and continued consumption for 13 weeks. The results revealed that this synthetic diet altered locomotor activity. Additionally, by sequencing the 16S rRNA gene from fecal samples (collected weekly) in each group, we identified a decrease in bacterial diversity, and through PICRUSt2 analysis, we predicted changes in metabolic pathway activities. Furthermore, we saw a decrease in the production of short chain fatty acids and a shortening of the colon in mice consuming the synthetic diet. Finally, we measured TNF-α, IL-6, and IL-10 in serum, the hippocampus, and colon, and found that the synthetic diet significantly increased IL-6 production in the hippocampus. These results demonstrate the importance of a multidisciplinary approach to future diet and microbiome studies, as diet not only impacts the gut microbiome, but potentially cognitive and immune function as well.
- Micro SpectrumSupragingival mycobiome of HIV-exposed-but-uninfected children reflects a stronger correlation with caries-free associated taxa compared to HIV-infected or uninfected childrenL. O’Connell , A.E. Mann, E. Osagie , and 8 more authorsMicrobiology Spectrum, 2023
Highly active antiretroviral treatment (HAART) has greatly reduced opportunistic infections in HIV-infected individuals. However, even with use of HAART, HIV-infected individuals are still at an increased risk of oral diseases, including dental caries. Dental caries development is associated with microbial community shifts leading to community dysbiosis. HIV infection can significantly alter the bacteriome and mycobiome composition in the oral cavity; however, these impacts have not been assessed for caries initiation and progression. The aim of this study was to characterize the mycobiome for supragingival plaque samples from HIV-infected (HI), HIV-exposed-but-uninfected (HEU), and HIV-unexposed-and-uninfected (HUU) children with and without caries. To accomplish this, ITS1 amplicons of 127 samples were sequenced. We found that HIV infection and exposure resulted in changes to the supragingival plaque mycobiome for both health and caries. Overall, a reduction in community diversity as caries progressed was observed, with HI children having the lowest diversity and HEU children the highest. \emphCandida albicans was the most abundant species identified, with 177 different ASVs. Two \emphC. albicans ASVs dominated the data (53.0% and 38.5% of all taxonomic assignments). The more frequent ASV dominated HI and HUU communities, whereas the second dominated HEU communities. HEU children also had the lowest abundance of \emphC. albicans and the highest number of health-associated taxa.
- µLifeThe eukaryome of African children is influenced by geographic location, gut biogeography, and nutritional status.P. Vonaesch , V. Billy , A.E. Mann, and 7 more authorsmicroLife, 2023
Eukaryotes have historically been studied as parasites but recent evidence suggests they may be indicators of a healthy gut ecosystem. Here, we describe the eukaryome along the gastrointestinal tract of children 2-5 years and test for associations with clinical factors such as anaemia, intestinal inflammation, chronic undernutrition and age. Children were enrolled from December 2016 - May 2018 in Bangui, Central African Republic and Antananarivo, Madagascar. We analyzed a total of 1,104 samples representing 212 gastric, 187 duodenal, and 705 fecal samples using a metabarcoding approach targeting the full ITS2 region for fungi, and the V4 hypervariable region of the 18S rRNA gene for the overall eukaryome. Roughly half of all fecal samples showed microeukaryotic reads. We find high inter-subject variability, only a handful of taxa that are likely residents of the gastrointestinal tract, and frequent co-occurrence of eukaryotes within an individual. We also find that the eukaryome differs between the stomach, duodenum and feces and is strongly influenced by country of origin. Our dat ashow trends toward higher levels of \emphFusarium equiseti, a mycotoxin producing fungus, and lower levels of the protist \emphBlastocystis in stunted chiuldren compared to non-stunted controls. Overall, the eukaryome is poorly correlated with clinical variables. Our study is of one of the largest cohorts analyzing the human intestinal eukaryome to date and the first to compare the eukaryome across different compartments of the gastrointestinal tract. Our results highlight the importance of studying populations across the world to uncover common features of the eukaryome in health.
-
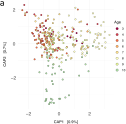 Impact of HIV on the Oral Microbiome of Children Living in Sub-Saharan Africa, Determined by Using an \emphrpoC Gene Fragment Metataxonomic Approach.A.E. Mann, L.M. O’Connell , E. Osagie , and 6 more authorsMicrobiology Spectrum, 2023
Impact of HIV on the Oral Microbiome of Children Living in Sub-Saharan Africa, Determined by Using an \emphrpoC Gene Fragment Metataxonomic Approach.A.E. Mann, L.M. O’Connell , E. Osagie , and 6 more authorsMicrobiology Spectrum, 2023Children living with HIV have a higher prevalence of oral diseases including caries, the mechanisms of which are not well understood. Here, we test the hypothesis that HIV infection is associated with a more cariogenic oral microbiome, characterized by an increase in bacteria involved in the pathogenesis of caries. We present data generated from supragingival plaques collected from 484 children representing three exposure groups: [1] children living with HIV (HI), [2] children who were perinatally exposed but uninfected (HEU), and [3] unexposed and therefore uninfected children (HUU). We found that the microbiome of HI children is distinct as compared to HEU or HUU children, and that this distinction is more pronounced in diseased teeth as compared to healthy teeth, suggesting that the impact of HIV is more severe as caries progresses. Moreover, we report both an increase in bacterial diversity and decrease in community similarity in our older as compared to younger HI cohort, which may in part be a prolonged effect of HIV and/or its treatment. Finally, while Streptococcus mutans is often a dominant species in late-stage caries, it tends to be found at lower frequency in our HI cohort as compared to other groups. Our results highlight the taxonomic diversity of the supragingival plaque microbiome and suggest that broad and increasingly individualistic ecological shifts are responsible for the pathogenesis of caries in children living with HIV, coupled with a diverse and possibly severe impact on known cariogenic taxa that potentially exacerbates caries.
-
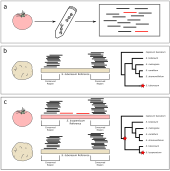 Do I have something in my teeth? The trouble with genetic analyses of diet from archaeological dental calculusA.E. Mann, J.A. Fellows Yates , Z. Fagernäs , and 3 more authorsQuaternary International, 2023
Do I have something in my teeth? The trouble with genetic analyses of diet from archaeological dental calculusA.E. Mann, J.A. Fellows Yates , Z. Fagernäs , and 3 more authorsQuaternary International, 2023Dental calculus and other preserved microbiome substrates are an attractive target for dietary reconstruction in past populations through a variety of physical, chemical, and molecular means. Recently, studies have attempted to reconstruct diet from archaeological dental calculus using archaeogenetic techniques. While dental calculus may provide a relatively stable environment for DNA preservation, the detection of plants and animals possibly consumed by an individual through DNA analysis is primarily hindered by microbial richness and incomplete reference databases. Moreover, high genomic similarity within eukaryotic groups - such as mammals - can obfuscate precise taxonomic identification. In the current study we demonstrate the challenges associated with accurate taxonomic identification and authentication of dietary taxa in ancient DNA data using both synthetic and ancient dental calculus datasets. We highlight common errors and sources of contamination across ancient DNA datasets, provide recommendations for dietary DNA validation, and call for caution in the interpretation of diet from dental calculus and other archaeological microbiome substrates.


2022
-
 The Devil is in the Details: Variable impacts of season, BMI, sampling site temperature, and presence of insects on the post-mortem microbiomeA.M. Tarone , A.E. Mann, Y. Zhang , and 5 more authorsFrontiers in Microbiology, 2022
The Devil is in the Details: Variable impacts of season, BMI, sampling site temperature, and presence of insects on the post-mortem microbiomeA.M. Tarone , A.E. Mann, Y. Zhang , and 5 more authorsFrontiers in Microbiology, 2022Post-mortem microbial communities are increasingly investigated as proxy evidence for a variety of factors of interest in forensic science. The reported predictive power of the microbial community to determine aspects of the individual’s post-mortem history (e.g., the post-mortem interval) varies substantially among published research. This observed variation is partially driven by the local environment or the individual themselves. In the current study, we investigated the impact of BMI, sex, insect activity, season, repeat sampling, decomposition time, and temperature on the microbial community sampled from donated human remains in San Marcos, TX using a high-throughput gene-fragment metabarcoding approach. We found that season, temperature at the sampling site, BMI, and sex had a significant effect on the post-mortem microbiome, the presence of insects has a homogenizing influence on the total bacterial community, and that community consistency from repeat sampling decreases as the decomposition process progresses. Moreover, we demonstrate the importance of temperature at the site of sampling on the abundance of important diagnostic taxa. The results of this study suggest that while the bacterial community or specific bacterial species may prove to be useful for forensic applications, a clearer understanding of the mechanisms underpinning microbial decomposition will greatly increase the utility of microbial evidence in forensic casework.

2021
- BMC Oral HealthDental caries and its association with the oral microbiomes and HIV in young children - Nigeria (DOMHaIN): a cohort studyM.O. Coker , P. Akhigbe , E. Osagie , and 10 more authorsBMC Oral Health, 2021
Background: This study seeks to understand better the mechanisms underlying the increased risk of caries in HIV-infected school-aged Nigerian children by examining the relationship between the plaque microbiome and perinatal HIV infection and exposure. We also seek to investigate how perinatal HIV infection and exposure impact tooth-specific microbiomes’ role on caries disease progression. Methods: The participants in this study were children aged 4 to 11 years recruited from the University of Benin Teaching Hospital (UBTH), Nigeria, between May to November 2019. Overall, 568 children were enrolled in three groups: 189 HIV-infected (HI), 189 HIV-exposed but uninfected (HEU) and 190 HIV-unexposed and uninfected (HUU) as controls at visit 1 with a 2.99% and 4.90% attrition rate at visit 2 and visit 3 respectively. Data were obtained with standardized questionnaires. Blood samples was collected for HIV, HBV and HCV screening; CD4, CD8 and Full Blood Count analysis; and plasma samples stored for future investigations; oral samples including saliva, buccal swabs, oropharyngeal swab, tongue swab, dental plaque were collected aseptically from participants at different study visits. Conclusions: Results from the study will provide critical information on how HIV exposure, infection, and treatment, influence the oral microbiome and caries susceptibility in children. By determining the effect on community taxonomic structure and gene expression of dental microbiomes, we will elucidate mechanisms that potentially create a predisposition for developing dental caries. As future plans, the relationship between respiratory tract infections, immune and inflammatory markers with dental caries in perinatal HIV infection and exposure will be investigated.
- PNASThe evolution and changing ecology of the African hominid oral microbiomeJ.A. Fellows Yates , I.M. Velsko , F. Aron , and 46 more authorsProceedings of the National Academy of Sciences, 2021
The oral microbiome plays key roles in human biology, health, and disease, but little is known about the global diversity, variation, or evolution of this microbial community. To better understand the evolution and changing ecology of the human oral microbiome, we analysed 124 dental biofilm metagenomes from humans, including Neanderthals and Late Pleistocene to present-day modern humans, chimpanzees, and gorillas, as well as New World howler monkeys for comparison. We find that a core microbiome of primarily biofilm-structural taxa has been maintained throughout African hominid evolution, and these microbial groups are also shared with howler monkeys, suggesting that they have been important oral members since before the catarrhine-platyrrhine split ca. 40 million years ago. However, community structure and individual microbial phylogenies do not closely reflect host relationships, and the dental biofilms of Homoand chimpanzees are distinguished by major taxonomic and functional differences. Reconstructing oral metagenomes up to 100 thousand years ago, we show that the microbial profiles of both Neanderthals and modern humans are highly similar, sharing functional adaptations in nutrient metabolism. These include an apparent Homo-specific acquisition of salivary amylase-binding capability by oral streptococci, suggesting microbial co-adaptation with host diet. We additionally find evidence of shared genetic diversity in the oral bacteria of Neanderthal and Upper Palaeolithic modern humans that is not observed in later modern human populations. Differences in the oral microbiomes of African hominids provide insights into human evolution, the ancestral state of the human microbiome, and a temporal framework for understanding microbial health and disease.
- Microorg.The adult phenylketonuria (PKU) gut microbiomeV.J. Mancilla , A.E. Mann, Y. Zhang , and 1 more authorMicroorganisms, 2021
Phenylketonuria (PKU) is an inborn error of phenylalanine metabolism primarily treatedthrough a phenylalanine-restrictive diet that is frequently supplemented with an amino acid formulato maintain proper nutrition. Little is known of the effects of these dietary interventions on thegut microbiome of PKU patients, particularly in adults. In this study, we sequenced the V4 regionof the 16S rRNA gene from stool samples collected from adults with PKU (n= 11) and non-PKUcontrols (n= 21). Gut bacterial communities were characterized through measurements of diversityand taxa abundance. Additionally, metabolic imputation was performed based on detected bacteria.Gut community diversity was lower in PKU individuals, though this effect was only statisticallysuggestive. A total of 65 genera across 5 phyla were statistically differentially abundant betweenPKU and control samples (p< 0.001). Additionally, we identified six metabolic pathways that differedbetween groups (p< 0.05), with four enriched in PKU samples and two in controls. While the childPKU gut microbiome has been previously investigated, this is the first study to explore the gutmicrobiome of adult PKU patients. We find that microbial diversity in PKU children differs from PKUadults and highlights the need for further studies to understand the effects of dietary restrictions.
2020
-
 Comparison of the Bacterial Gut Microbiome of North American Trypanosoma spp. With and Without Trypanosoma cruziA.E. Mann, E.A. Mitchell , Y. Zhang , and 4 more authorsFrontiers in Microbiology, 2020
Comparison of the Bacterial Gut Microbiome of North American Trypanosoma spp. With and Without Trypanosoma cruziA.E. Mann, E.A. Mitchell , Y. Zhang , and 4 more authorsFrontiers in Microbiology, 2020Chagas disease, caused by the hemoflagellate protist Trypanosoma cruzi, affects nearly 6 million people worldwide, mainly in Latin America. Hematophagous triatomine insects (“kissing bugs”) are the primary vectors of T. cruzi throughout the Americas and feed on a variety of animals, including humans. Control of triatomines is central to the control of T. cruzi infection. Recent advances in mitigation of other insect-borne diseases via the manipulation of insect-associated bacteria as a way to halt or slow disease transmission has opened questions to the applicability of these methods to Chagas disease vectors. Few studies have examined the hindgut microbiome of triatomines found in North America. In the current study, two species of triatomines were collected across Texas, United States, screened for the presence of T. cruzi, and analyzed for the bacterial composition of their hindguts using a 16S rRNA gene-fragment metabarcoding approach. We compared diversity of microbial community profiles across 74 triatomine insects to address the hypothesis that the richness and abundance of bacterial groups differ by T. cruzi infection and strain type, blood meal engorgement status, insect species, sex, and collection location. The gut microbial community of individual triatomines was characterized by low intraindividual taxonomic diversity and high interindividual variation that was weakly predicted by triatomine species, and was not predicted by triatomine sex, collection location, T. cruzi infection status, or blood meal score. However, we did find bacterial groups enriched in T. cruzi-positive individuals, including Enterobacterales, and Petrimonas. Additionally, we detected Salmonella enterica subspecies diarizonae in three triatomine individuals; this species is commonly associated with reptiles and domesticated animals and is a pathogen of humans. These data suggest that Triatoma spp. in Texas have variable patterns of colonized and transient bacteria, and may aid in development of novel means to interfere with transmission of the Chagas disease parasite T. cruzi. Deeper understanding of the effects of parasite infection on diverse insect vector microbiomes may highlight disease transmission risk and facilitate discovery of possible intervention strategies for biological control of this emerging vector-borne disease of global health significance.
-
 Biodiversity of protists and nematodes in the wild non-human primate gutA.E. Mann, F. Mazel , M.A. Lemay , and 18 more authorsThe ISME Journal, 2020
Biodiversity of protists and nematodes in the wild non-human primate gutA.E. Mann, F. Mazel , M.A. Lemay , and 18 more authorsThe ISME Journal, 2020Documenting the natural diversity of eukaryotic organisms in the nonhuman primate (NHP) gut is important for understanding the evolution of the mammalian gut microbiome, its role in digestion, health and disease, and the consequences of anthropogenic change on primate biology and conservation. Despite the ecological significance of gut-associated eukaryotes, little is known about the factors that influence their assembly and diversity in mammals. In this study, we used an 18S rRNA gene fragment metabarcoding approach to assess the eukaryotic assemblage of 62 individuals representing 16 NHP species. We find that cercopithecoids, and especially the cercopithecines, have substantially higher alpha diversity than other NHP groups. Gut-associated protists and nematodes are widespread among NHPs, consistent with their ancient association with NHP hosts. However, we do not find a consistent signal of phylosymbiosis or host-species specificity. Rather, gut eukaryotes are only weakly structured by primate phylogeny with minimal signal from diet, in contrast to previous reports of NHP gut bacteria. The results of this study indicate that gut-associated eukaryotes offer different information than gut-associated bacteria and add to our understanding of the structure of the gut microbiome.


2019
- AJPAThe efficacy of whole genome capture on ancient dental calculus and dentinK.A. Ziesemer , J. Ramos-Madrigal , A.E. Mann, and 14 more authorsAmerican Journal of Physical Anthropology, 2019
Dental calculus is among the richest known sources of ancient DNA in the archaeological record. Although most DNA within calculus is microbial, it has been shown to contain sufficient human DNA for the targeted retrieval of whole mitochondrial genomes. Here, we explore whether calculus is also a viable substrate for whole human genome recovery using targeted enrichment techniques. Total DNA extracted from 24 paired archaeological human dentin and calculus samples was subjected to whole human genome enrichment using in‐solution hybridization capture and high‐throughput sequencing. Total DNA from calculus exceeded that of dentin in all cases, and although the proportion of human DNA was generally lower in calculus, the absolute human DNA content of calculus and dentin was not significantly different. Whole genome enrichment resulted in up to four‐fold enrichment of the human endogenous DNA content for both dentin and dental calculus libraries, albeit with some loss in complexity. Recovering more on‐target reads for the same sequencing effort generally improved the quality of downstream analyses, such as sex and ancestry estimation. For nonhuman DNA, comparison of phylum‐level microbial community structure revealed few differences between precapture and postcapture libraries, indicating that off‐target sequences in human genome‐enriched calculus libraries may still be useful for oral microbiome reconstruction. While ancient human dental calculus does contain endogenous human DNA sequences, their relative proportion is low when compared with other skeletal tissues. Whole genome enrichment can help increase the proportion of recovered human reads, but in this instance enrichment efficiency was relatively low when compared with other forms of capture. We conclude that further optimization is necessary before the method can be routinely applied to archaeological samples.
2018
-
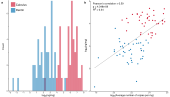 Differential preservation of endogenous human and microbial DNA in dental calculus and dentinA.E. Mann, S. Sabin , K.A. Ziesemer , and 15 more authorsScientific Reports, 2018
Differential preservation of endogenous human and microbial DNA in dental calculus and dentinA.E. Mann, S. Sabin , K.A. Ziesemer , and 15 more authorsScientific Reports, 2018Dental calculus (calcified dental plaque) is prevalent in archaeological skeletal collections and is a rich source of oral microbiome and host-derived ancient biomolecules. Recently, it has been proposed that dental calculus may provide a more robust environment for DNA preservation than other skeletal remains, but this has not been systematically tested. In this study, shotgun-sequenced data from paired dental calculus and dentin samples from 48 globally distributed individuals are compared using a metagenomic approach. Overall, we find DNA from dental calculus is consistently more abundant and less contaminated than DNA from dentin. The majority of DNA in dental calculus is microbial and originates from the oral microbiome; however, a small but consistent proportion of DNA (mean 0.08 ± 0.08%, range 0.007–0.47%) derives from the host genome. Host DNA content within dentin is variable (mean 13.70 ± 18.62%, range 0.003–70.14%), and for a subset of dentin samples (15.21%), oral bacteria contribute > 20% of total DNA. Human DNA in dental calculus is highly fragmented, and is consistently shorter than both microbial DNA in dental calculus and human DNA in paired dentin samples. Finally, we find that microbial DNA fragmentation patterns are associated with guanine-cytosine (GC) content, but not aspects of cellular structure.

2017
-
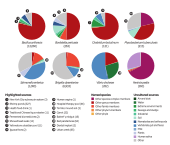 A robust framework for microbial archaeologyC. Warinner , A. Herbig , A.E. Mann, and 5 more authorsAnnual Review of Genomics and Human Genetics, 2017
A robust framework for microbial archaeologyC. Warinner , A. Herbig , A.E. Mann, and 5 more authorsAnnual Review of Genomics and Human Genetics, 2017Microbial archaeology is flourishing in the era of high-throughput sequencing, revealing the agents behind devastating historical plagues, identifying the cryptic movements of pathogens in prehistory, and reconstructing the ancestral microbiota of humans. Here, we introduce the fundamental concepts and theoretical framework of the discipline, then discuss applied methodologies for pathogen identification and microbiome characterization from archaeological samples. We give special attention to the process of identifying, validating, and authenticating ancient microbes using high-throughput DNA sequencing data. Finally, we outline standards and precautions to guide future research in the field.

2015
- Sci. Rep.Intrinsic challenges in ancient microbiome reconstruction using 16S rRNA gene amplificationK.A. Ziesemer , A.E. Mann, K. Sankaranarayanan , and 17 more authorsScientific Reports, 2015
To date, characterization of ancient oral (dental calculus) and gut (coprolite) microbiota has been primarily accomplished through a metataxonomic approach involving targeted amplification of one or more variable regions in the 16S rRNA gene. Specifically, the V3 region (E. coli 341–534) of this gene has been suggested as an excellent candidate for ancient DNA amplification and microbial community reconstruction. However, in practice this metataxonomic approach often produces highly skewed taxonomic frequency data. In this study, we use non-targeted (shotgun metagenomics) sequencing methods to better understand skewed microbial profiles observed in four ancient dental calculus specimens previously analyzed by amplicon sequencing. Through comparisons of microbial taxonomic counts from paired amplicon (V3 U341F/534R) and shotgun sequencing datasets, we demonstrate that extensive length polymorphisms in the V3 region are a consistent and major cause of differential amplification leading to taxonomic bias in ancient microbiome reconstructions based on amplicon sequencing. We conclude that systematic amplification bias confounds attempts to accurately reconstruct microbiome taxonomic profiles from 16S rRNA V3 amplicon data generated using universal primers. Because in silico analysis indicates that alternative 16S rRNA hypervariable regions will present similar challenges, we advocate for the use of a shotgun metagenomics approach in ancient microbiome reconstructions.